Resumen Breve
Este video ofrece una visión general de las neoplasias mieloproliferativas crónicas (NMP), un grupo heterogéneo de enfermedades clonales de la línea mieloide. Se discuten la clasificación, el contexto histórico, la etiología, el cuadro clínico, el diagnóstico de laboratorio y el tratamiento de estas neoplasias, con un enfoque en la leucemia mieloide crónica (LMC), la policitemia vera (PV), la trombocitemia esencial (TE) y la mielofibrosis primaria (MFP).
- Las NMP son enfermedades clonales de la línea mieloide que resultan en la proliferación excesiva de células mieloides en la médula ósea, la sangre periférica y los tejidos.
- La clasificación de la OMS de 2022 se basa en criterios clínicos, histológicos y moleculares.
- Las mutaciones genéticas, como JAK2, CALR y MPL, desempeñan un papel importante en la patogénesis de las NMP.
- El tratamiento ha evolucionado desde soluciones de Fowler y busulfano hasta inhibidores de la tirosina quinasa (ITK) como imatinib, nilotinib y dasatinib.
Introducción a las Neoplasias Mieloproliferativas Crónicas
Las neoplasias mieloproliferativas crónicas (NMP) son un grupo diverso de enfermedades clonales que afectan la línea mieloide. Estas enfermedades se originan en la célula madre hematopoyética en la médula ósea, lo que lleva a una proliferación excesiva y acumulación de células mieloides sin un bloqueo en la maduración o función. Las NMP se clasifican según la línea celular predominante afectada, ya sea eritroide, granulocítica o plaquetaria. Estas neoplasias se caracterizan por una evolución crónica, esplenomegalia, hiperplasia medular trilineal y recuentos sanguíneos elevados, con eosinofilia y basofilia frecuentes. Además, pueden presentar hematopoyesis extramedular, tendencia a la fibrosis y complicaciones hemorrágicas o trombóticas, así como alteraciones moleculares responsables del fenotipo mieloproliferativo.
Clasificación y Contexto Histórico
La clasificación actual de las NMP, según la Organización Mundial de la Salud (OMS) en su revisión de 2022, se basa en criterios clínicos, histológicos y moleculares. Las NMP más relevantes en la práctica clínica son la leucemia mieloide crónica (LMC), que es cromosoma Filadelfia positivo, y las neoplasias mieloproliferativas crónicas cromosoma Filadelfia negativas clásicas, que incluyen la policitemia vera (PV), la trombocitemia esencial (TE), la mielofibrosis primaria (MFP), la leucemia crónica neutrofílica, la leucemia crónica eosinofílica, la leucemia mielomonocítica juvenil y las neoplasias mieloproliferativas no especificadas. Históricamente, la LMC fue descrita entre 1939 y 1945, la mielofibrosis primaria en 1879, la policitemia vera en 1892 y la trombocitemia esencial en 1934. En 1951, William Dameshek acuñó el término "síndrome mieloproliferativo" para englobar estas patologías y describir sus semejanzas clínicas.
Descubrimiento del Cromosoma Filadelfia y Mutación JAK2
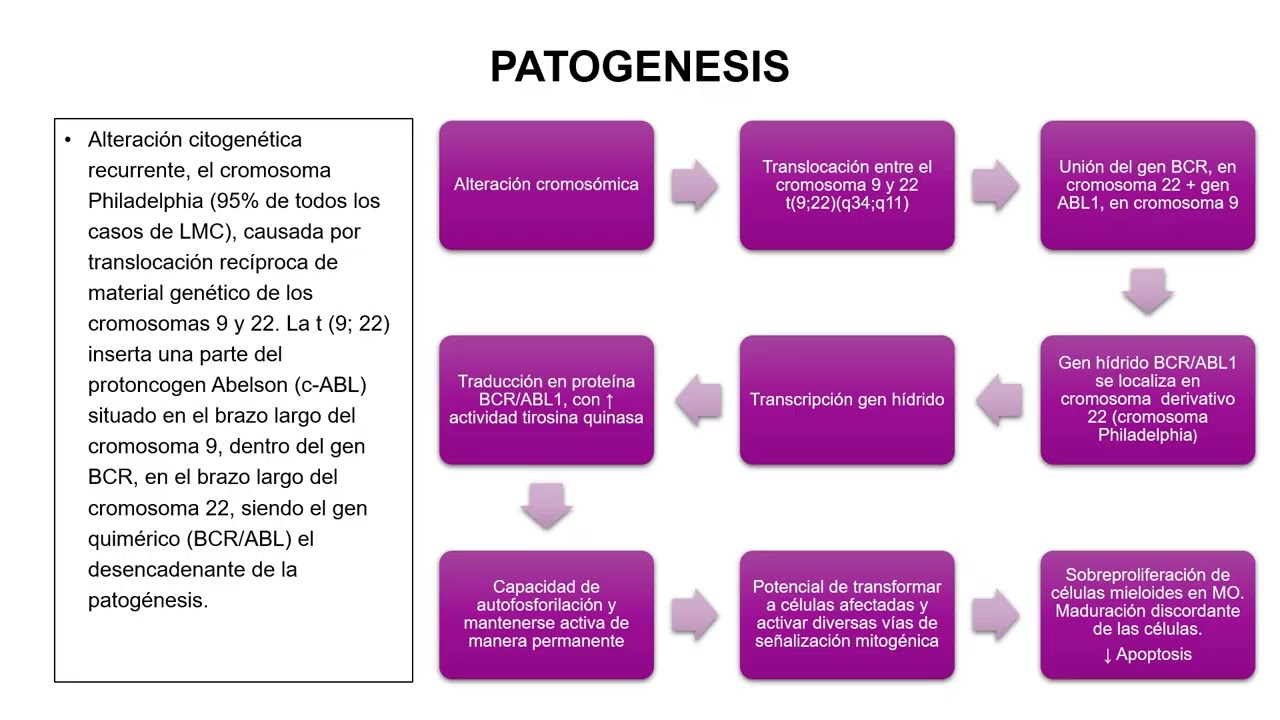
El cromosoma Filadelfia, descubierto en 1960, es el resultado de una traslocación recíproca entre los cromosomas 9 y 22, designada t(9;22)(q34;q11). Esta traslocación da como resultado la fusión del gen BCR con el gen ABL, creando un gen quimérico BCR-ABL que codifica una proteína con actividad tirosina quinasa constitutiva. Esta actividad tirosina quinasa es responsable de la activación de otras vías de transducción de señales que promueven la proliferación celular, reducen la adhesión celular y disminuyen la apoptosis, características del fenotipo leucémico en la LMC. En 2005, se identificó la mutación de la quinasa Janus 2 (JAK2), confirmando el carácter clonal de las neoplasias mieloproliferativas y su inclusión en la clasificación de la OMS en 2008. La mutación JAK2, localizada en el exón 14 del gen JAK2, induce la activación constitutiva de JAK2 y de las vías de señalización intracelular, siendo la alteración molecular más frecuente en las NMP, presente en más del 95% de los pacientes con PV y en el 50-60% de aquellos con TE y MFP.
Leucemia Mieloide Crónica (LMC): Etiología y Patogenia
La leucemia mieloide crónica (LMC) resulta de una mutación somática en una célula hematopoyética multipotente, lo que lleva a la proliferación clonal y generación de células mieloides granulocíticas que mantienen su capacidad de diferenciación. La LMC tiene una evolución crónica que, sin tratamiento adecuado, puede progresar a una fase aguda o terminal similar a una leucemia aguda. Se caracteriza por la presencia de células normales y diferenciadas en la médula ósea y la sangre periférica, en contraste con las leucemias agudas, donde hay una detención en la maduración. La etiología de la LMC es desconocida, pero hay un aumento de la incidencia tras la exposición a radiaciones ionizantes o agentes químicos como el benceno. La presencia del cromosoma Filadelfia confirma que la LMC es una enfermedad clonal que resulta de la transformación maligna de una célula progenitora pluripotencial hematopoyética.
Mecanismo Molecular y Cuadro Clínico de la LMC
El cromosoma Filadelfia es un cromosoma 22 disminuido de tamaño debido a una traslocación recíproca con el cromosoma 9, designada t(9;22)(q34;q11). Esta traslocación resulta en la creación de un gen quimérico BCR-ABL que codifica una proteína de fusión con actividad tirosina quinasa constitutiva. Esta actividad tirosina quinasa es responsable de la activación de otras vías de transducción de señales que promueven la proliferación celular, reducen la adhesión celular y disminuyen la apoptosis. Clínicamente, entre el 20 y el 40% de los pacientes con LMC son asintomáticos al momento del diagnóstico, y la alteración se descubre por la detección de leucocitosis en una analítica de rutina. Los signos y síntomas de la enfermedad son de aparición paulatina y resultan de la anemia y la esplenomegalia. Los pacientes pueden experimentar hepatoesplenomegalia, dolor o plenitud en el cuadrante superior izquierdo abdominal, dolor óseo, artritis gotosa, hiperviscosidad sanguínea y tendencia a la trombosis.
Diagnóstico de Laboratorio y Evolución de la LMC
En el laboratorio, la LMC se caracteriza por una leucocitosis intensa, que va desde 50,000 hasta 800,000 leucocitos por milímetro cúbico, con desviación a la izquierda y presencia de elementos inmaduros de la serie mieloide. La presencia y el conteo de blastos definen la fase de la enfermedad. La evolución de la LMC muestra un curso evolutivo de dos etapas: una fase crónica y una crisis blástica o fase aguda. En la fase crónica, los pacientes tienen una clínica oligosintomática con quejas inespecíficas y leucocitosis en sangre periférica, generalmente superior a 25,000 células, con granulocitos en todas sus etapas de maduración. En la crisis blástica o fase aguda, se observa la presencia de más del 20% de blastos en sangre periférica o médula ósea, o infiltración extramedular de blastos.
Fases de la LMC y Diagnóstico Diferencial
En la fase crónica de la LMC, se observa leucocitosis con granulocitos en todas sus etapas de maduración, pudiendo encontrar blastos, lo que se denomina desviación a la izquierda escalonada con maduración conservada. Un hallazgo clásico es el hiato leucémico, donde hay más mielocitos que metamielocitos. La basofilia y eosinofilia pueden encontrarse en casi el 90% de los casos, y puede haber monocitosis. La anemia es discreta o moderada, normocítica, normocrómica, con un variado grado de anisocitosis, pero sin reticulocitosis. La médula es hipercelular con disminución de tejido adiposo e hiperplasia de la serie granulocítica. En el 2022, se redefinió la LMC como una enfermedad bifásica con una fase crónica y una crisis blástica o fase aguda. Es importante realizar un diagnóstico diferencial con las reacciones leucemoides, que se caracterizan por la ausencia de esplenomegalia, escasa o nula mielemia, ausencia de basofilia, presencia de granulaciones tóxicas y cuerpos de Döhle, y aumento de la fosfatasa alcalina leucocitaria.
Tratamiento de la LMC: Evolución Histórica e Inhibidores de la Tirosina Quinasa
Históricamente, el tratamiento de la LMC ha evolucionado desde la solución de Fowler (arsenito potásico) y el busulfano hasta los inhibidores de la tirosina quinasa (ITK). A fines de la década de 1990, apareció el inhibidor de las transducciones de señales número 571, que inhibe la autofosforilación de las quinasas de ABL, conocido como imatinib. Posteriormente, se describió la resistencia a imatinib, lo que llevó al desarrollo de inhibidores de segunda generación como nilotinib y dasatinib. Los ITK de primera generación (imatinib) y de segunda generación (dasatinib y nilotinib) son inhibidores selectivos de los ITK mutantes e inducen remisión hematológica en el 98% de los pacientes y remisión citogenética en al menos el 80%, siendo actualmente el tratamiento de primera línea para la LMC. Las opciones de tratamiento dependen del estadio de la enfermedad, la edad del paciente, las comorbilidades, los factores pronósticos y la disponibilidad de un donante de médula ósea compatible.
Respuestas al Tratamiento y Concepto de Hematopoyesis
Después de iniciar el tratamiento con ITK, se espera una respuesta que puede ser hematológica completa (leucocitos < 10,000, plaquetas < 450,000, ausencia de mielemia circulante y desaparición de síntomas), citogenética mayor (menos del 35% de células Filadelfia positivas), citogenética completa (ausencia de células Filadelfia positivas), molecular mayor (menos de 0.05% de transcrito BCR-ABL) o molecular completa (transcrito BCR-ABL indetectable). El concepto de hematopoyesis es fundamental para entender las NMP. La célula madre hematopoyética se diferencia en célula madre mieloide y célula madre linfoide. De la célula madre mieloide se originan los glóbulos rojos, los granulocitos, los monocitos y las plaquetas. Las NMP se caracterizan por una proliferación patológica clonal maligna de la línea mieloide, lo que puede resultar en policitemia vera (proliferación de glóbulos rojos), leucemia mieloide crónica (proliferación de granulocitos con cromosoma Filadelfia) o trombocitemia esencial (proliferación de megacariocitos con aumento de plaquetas).
Policitemia Vera: Etiología y Fisiopatología
La policitemia vera (PV) es una enfermedad clonal maligna del sistema hematopoyético con proliferación principal del sector eritrocitario, pero también de los sectores granulocítico y megacariocítico. La manifestación más prominente es un aumento de la masa de glóbulos rojos y una elevación persistente del hematocrito. La PV se desarrolla a partir de la transformación de una única célula madre hematopoyética con la ventaja de un crecimiento selectivo que se transforma gradualmente en el principal precursor mieloide. Las colonias eritroides derivadas de la médula ósea se desarrollan en ausencia de eritropoyetina endógena, lo que revela el carácter autosómico de la proliferación. La PV es fisiopatológicamente distinta de las eritrocitosis secundarias, que generalmente ocurren debido a niveles elevados de eritropoyetina. En la PV, el JAK2 está activado sin necesidad de eritropoyetina debido a una mutación que causa la activación constitutiva del JAK2.
Criterios Diagnósticos y Manifestaciones Clínicas de la Policitemia Vera
Los criterios diagnósticos de la PV, según la Organización Mundial de la Salud (OMS), incluyen criterios mayores (hemoglobina elevada, mielosis con megacariocitos pleomorfos y mutación JAK2) y criterios menores (niveles séricos disminuidos de eritropoyetina). El diagnóstico requiere el cumplimiento de los tres criterios mayores o de dos criterios mayores más el criterio menor. Las manifestaciones clínicas de la PV están asociadas al aumento de la viscosidad sanguínea y a la eritropoyesis patológica. Los síntomas neurológicos incluyen cefaleas, mareos, trastornos visuales y parestesias. Otros síntomas incluyen prurito acuogénico, crisis de gota, plétora facial, esplenomegalia, hepatomegalia e hipertensión. En el laboratorio, se observa aumento de la hemoglobina, hematocrito y glóbulos rojos, leucocitosis, trombocitosis, mutación JAK2, médula ósea hipercelular, eritropoyetina sérica baja, aumento del ácido úrico y LDH, y presencia de anormalidades cromosómicas.
Trombocitemia Esencial: Etiología y Cuadro Clínico
La trombocitemia esencial (TE) es una neoplasia que se caracteriza por hiperplasia megacariocítica en la médula ósea, lo que resulta en un incremento persistente de las cifras de plaquetas, generalmente mayores a 600,000. La TE tiene un curso clínico relativamente benigno, pero puede presentar complicaciones trombóticas, arteriales y venosas, así como hemorrágicas. Entre un 40 y 50% de los pacientes son asintomáticos al momento del diagnóstico. Los eventos trombóticos pueden incluir isquemia cerebral transitoria, accidente cerebrovascular (ACV), oclusión de la vena porta o arteria retiniana, infarto agudo de miocardio, embolia pulmonar y trombosis venosa profunda. Los eventos hemorrágicos son menos frecuentes y pueden ser espontáneos o postraumáticos, con sangrado cutáneo, mucoso o digestivo.
Genética y Diagnóstico de la Trombocitemia Esencial
La trombocitemia esencial (TE) se caracteriza por una mayor sensibilidad de las células hematopoyéticas comprometidas a sus respectivos factores de crecimiento humorales primarios. Se han descubierto tres mutaciones somáticas importantes: JAK2, calreticulina (CALR) y el virus de la leucemia mieloproliferativa (MPL). Estas mutaciones conducen a una señalización aberrante independiente de citoquinas a través de la activación constitutiva de la vía JAK-STAT. La mutación JAK2 se encuentra en un 50-60% de los pacientes, las mutaciones CALR en un 15-25% y las mutaciones MPL alrededor del 3%. Los criterios diagnósticos de la OMS incluyen recuentos de plaquetas mayores a 450,000, criterios de médula ósea concordantes con trombocitemia, exclusión de otras patologías y demostración de la mutación JAK2, CALR o MPL.
Diagnóstico Diferencial y Características de Laboratorio de la Trombocitemia Esencial
Es importante tener en cuenta que la trombocitosis puede ser ocasionada por causas reactivas o procesos autónomos. Los procesos reactivos, también descritos como trombocitosis reactivas, son causados por procesos independientes al megacariocito y corresponden a la mayoría de los casos de trombocitosis en todas las edades y contextos clínicos. Entre las causas más comunes se incluyen el déficit de hierro, la hemorragia, hemólisis, infecciones virales, bacterianas, por micobacterias o fúngicas, procesos inflamatorios no infecciosos, patologías reumatológicas, trauma o reacciones medicamentosas o incluso en estado de posesplenectomía o asplenia funcional. En el hemograma de un paciente con TE, se pueden observar plaquetas elevadas, hemoglobina y hematocrito normales, leucocitosis moderada y basofilia. En el frotis, se observan agregados de plaquetas de morfología alterada, anisocitosis plaquetaria, plaquetas gigantes, vacuolización o hipogranularidad y fragmentos de citoplasma de megacariocitos circulantes.
Mielofibrosis Primaria: Características y Diagnóstico
La mielofibrosis primaria (MFP) se caracteriza por fibrosis de la médula ósea con hematopoyesis extramedular, esplenomegalia y anemia con dacriocitos y leucoeritroblastosis. El cuadro clínico puede incluir síntomas constitucionales como anorexia y síntomas B, prurito acuogénico y esplenomegalia. La tríada diagnóstica incluye esplenomegalia gigante, síndrome leucoeritroblástico con hematíes en lágrima y fibrosis medular. En el hemograma, se observa anemia normocítica normocrómica con anisocitosis y poiquilocitosis significativa, presencia de eritrocitos en lágrima o dacriocitos, leucopenia, leucocitosis y trombocitopenia. El mielograma generalmente se denomina seco debido al alto grado de fibrosis. La biopsia de médula ósea es imprescindible para el diagnóstico y puede mostrar distintos patrones de fibrosis.
Patogenia y Criterios Diagnósticos de la Mielofibrosis Primaria
La mielofibrosis es una hemopatía maligna originada en un progenitor hematopoyético clonal común a la serie mieloide y linfoide, en la cual la fibrosis de la médula ósea constituye un fenómeno secundario a una reacción de las células del microambiente medular como consecuencia de la liberación de citoquinas por parte de las células neoplásicas. La presencia o no de mutaciones en los tres genes, JAK2, CALR y MPL, se consideran cuatro genotipos distintos. El genotipo más frecuente es el JAK2, que representa en torno a un 60% de los casos, le sigue la mutación CALR, un 25% y las mutaciones MPL, que son bastante infrecuentes y solo se encuentran en el 5% de los casos. Los criterios diagnósticos de mielofibrosis incluyen criterios mayores (biopsia de médula ósea con proliferación de megacariocitos atípicos y fibrosis reticulínica o colágena, ausencia de evidencia de otros criterios de diagnóstico para otras neoplasias y demostración de las mutaciones) y criterios menores (anemia no achacable a otras causas o comorbilidades, leucocitosis, esplenomegalia palpable, aumento de la LDH y presencia del marcador clonal).
Leucemia Neutrofílica Crónica y Leucemia Eosinofílica Crónica
La leucemia neutrofílica crónica (LNC) se caracteriza por una proliferación granulocítica que implica leucocitosis con neutrofilia. Es poco frecuente y tiene un mal pronóstico, con una media de supervivencia de aproximadamente 2 años. La leucemia eosinofílica crónica (LEC) se caracteriza por un aumento de la proliferación de precursores y eosinófilos, lo que resulta en un aumento de eosinófilos anormales en sangre periférica y médula ósea, con infiltración en otros tejidos. Se debe hacer un diagnóstico diferencial de las causas de eosinofilia, como los desórdenes alérgicos, infecciones, parásitos, tumores y otras enfermedades autoinmunes.
Leucemia Mielomonocítica Juvenil y Resumen Comparativo de las NMP
La leucemia mielomonocítica juvenil (LMMJ) es una neoplasia agresiva de los niños caracterizada por una sobreproducción de monocitos que infiltran múltiples órganos. Es considerada como una sobreposición entre síndrome mielodisplásico y leucemia mieloproliferativa crónica por la OMS. El curso natural de la enfermedad es fatal, y el único tratamiento curativo es el trasplante. Finalmente, se presenta un cuadro comparativo de las principales características de la leucemia mieloide crónica, la policitemia vera, la trombocitemia esencial y la mielofibrosis primaria, incluyendo los niveles de eritrocitos, leucocitos, plaquetas, fibrosis, esplenomegalia, cromosoma Filadelfia y mutación JAK2.

